Overview – What is index hopping and what problem does it generate
Index hopping, also called index swapping or barcode mis-assignment is a phenomenon that occurs when multiplexed samples are sequenced on NGS platforms such as Illumina´s HiSeq using Exclusion-Amplification (ExAmp) chemistry. It has been observed that a certain amount of sequencing reads is incorrectly assigned from one sample to a different sample in a pool. Importantly, rates of index hopping dependent on the utilised library preparation method. The highest rates of index hopping have been found for PCR-free libraries and libraries that are contaminated with free adapters and primers. Even though the underlying mechanism remains somewhat elusive, it has been suggested that clean sequencing libraries are essential for sequencing on platforms such as the HiSeq or NovaSeq.
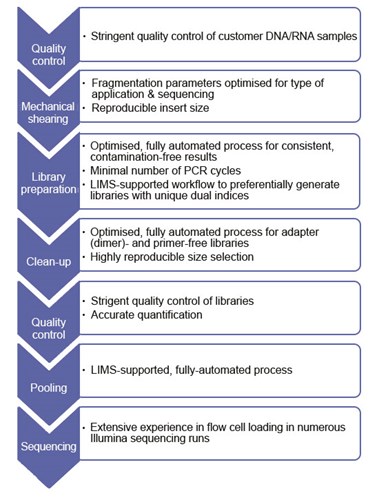
Workflow – Library preparation
Scientific expertise
Library preparation has the highest influence on levels of index hopping events. The library preparation process at Eurofins Genomics results in clean high-quality libraries with no detectable primer dimers or adapter dimers. We consequently have extremely low rates of index hopping.
At Eurofins Genomics, most steps of the library preparation are automated using liquid handlers and very strict purification steps are performed, which seem to mitigate this effect to nearly negligible amounts.
With our workflow, a non-uniquely dual indexed library may contain on average 0.008% of the reads coming from a library sharing one of the index sequences. This equals to 1 mis-assigned read per 1,250 correctly assigned reads. For example, if a non-uniquely dual indexed library was loaded with approximately 10% of total reads (e.g. 30 million read pairs) per lane and this library was affected by index hopping as another library present on the lane shared one of the indices, then 0.08% of reads (e.g. 24,000 read pairs) of the affected library would originate from the contaminating library.
Does this level of mis-assigned reads influence data interpretation?
For many study types such as whole genome sequencing and whole exome sequencing no influence is expected.
This includes re-sequencing projects that aim at detecting minor allele frequencies down to 1%, where usually a sequencing depth of 300x average coverage is recommended. This means that at least 3 unique reads with a specific mutation are needed in order to call a mutation. At 300x average coverage and an index hopping rate of 0.08% (experimental data-not shown), there is less than 30% chance that a single mis-assigned read with the mutation may be detected, which is well below the threshold of 3 mutated reads. Moreover, this will only be the case if the mutation is present at 100% in the “contaminating library”. If the mutation frequency is lower, the likelihood for carry-over is reduced even further! Therefore, rare mutation detection studies are very unlikely to be affected. If the contaminating library belongs to a different organism most of the index hopping reads will not map anyways, subsequently leaving the experimental data unaffected.
What is the effect of index hopping on RNA-Seq?
For RNA-Seq, where gene expression leves can vary substantially between sample types or treatments. The impact of index hopping, however, is very low in most cases. For example, if a cell line upregulates a certain transcript upon treatment the factor of 100 (e.g. from 10 FPKM to 1,000 FPKM), the index hopping could increase the FPKM (Fragments Per Kilobase Million) of the untreated control from 10 to 11 FPKM (~0.1% of 1000 FPKM). In conclusion, the fold change will not be substantially different.
Nevertheless, for single cell RNA-Seq, where commonly up to 384 libraries are pooled on a single lane, it is recommended to use uniquely indexed libraries in case very different cell types are analysed.
Eurofins Genomics also provides the fastest Sanger Sequencing services and optimised DNA and RNA oligonucleotides, such as PCR Primer NightXpress, SeqPrimer NightXpress, and SaltFree Oligo NightXpress.