Highlights
- High-Precision Variant Detection: Joint cohort calling minimizes false positives, enhances rare variant identification, and ensures consistent genotyping across samples.
- Robust Data Handling: Efficiently processes low-quality and low-depth data while reducing batch effects and inconsistencies.
- Comprehensive Analysis Workflow: Covers quality assessment, genome mapping, variant calling, annotation, and detailed reporting for reliable genomic insights.
- Advanced Biomarker Insights: Detects MSI status, TMB, CNV, and structural variations to support in-depth genomic research and clinical applications.
- Advanced algorithm: We use the advanced algorithm (Sentieon) to achieve highest accuracy and efficiency in variant detection. It enables precise germline variant calling, identifying SNPs and indels with reliability. The software ensures rapid processing of large-scale sequencing data, offering high throughput and scalability. Advanced quality control features, such as base quality score recalibration (BQSR) and duplicate marking, enhance data integrity. Additionally, it supports complex analyses like joint calling in cohort studies, improving rare and de novo variant detection. Optimized for speed and resource efficiency, Sentieon is ideal for large-scale genomic projects.
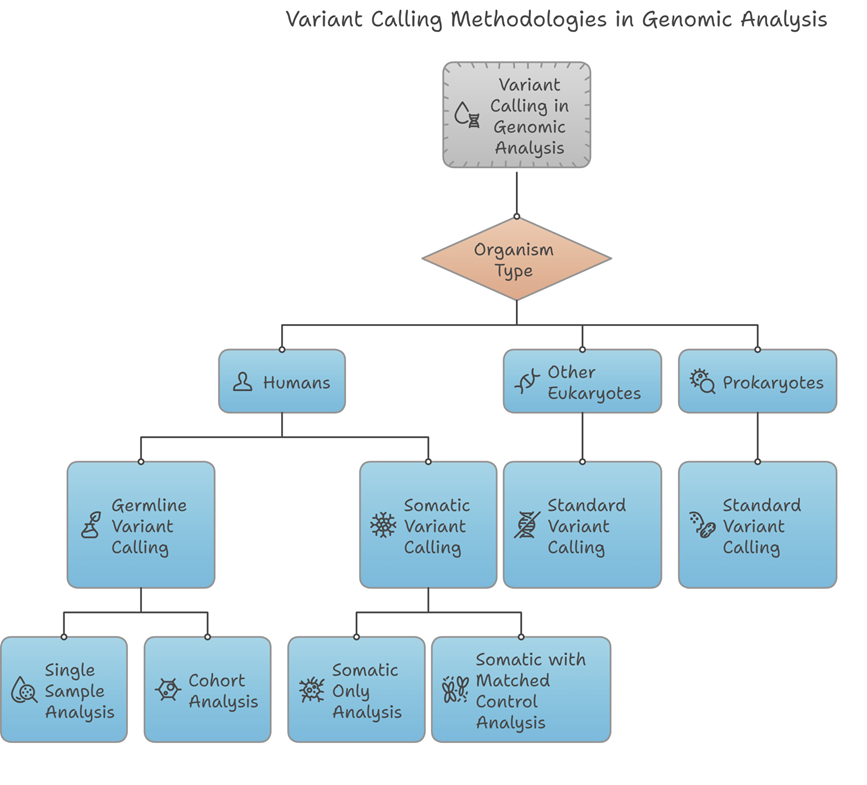
The diagram offers a structured approach to selecting the appropriate variant calling methodology based on organism type and analysis requirements.
For human samples, either Germline Variant Calling (for inherited mutations) or Somatic Variant Calling (for acquired mutations, such as those found in cancer) may be relevant. Germline analysis can be performed on a single sample or as part of a cohort study, while somatic analysis can be conducted independently or with a matched control to improve accuracy.
For other eukaryotic and prokaryotic organisms, a standard variant calling approach is applied.
This decision tree enables you to quickly identify the most suitable service based on your research focus and sample type, ensuring a highly relevant and efficient analysis.
Advantages of Joint Calling in a Cohort Study
1. Improved Variant Detection Accuracy
Joint calling considers all samples together, reducing false positives and improving sensitivity to rare variants.
2. Consistent Genotype Calling
Ensures uniform variant calling across all samples, minimizing batch effects and inconsistencies.
3. Better Handling of Low-Quality Data
Allows recovery of variants in low-depth regions by leveraging data from multiple samples.
4. Identification of Rare and De Novo Variants
Enhances the detection of rare mutations by comparing multiple individuals within the cohort.
5. More Robust Population-Level Analysis
Supports downstream analyses like allele frequency estimation and association studies.
Example: Joint Calling in Cancer Research
In a cancer genomics study, researchers analyze 100 patients with lung cancer to identify common and rare mutations. By performing joint variant calling, they detect shared mutations across multiple patients, improving confidence in variant calls. This helps in discovering novel cancer driver mutations, guiding targeted therapies and drug development.
Example: Joint Calling in Rare Genetic Disease Research
In a study on rare neurological disorders, researchers analyze whole exome sequencing data from 500 patients with an unknown inherited condition. By using joint variant calling, they can compare genetic variants across all patients and unaffected family members. This approach helps identify rare recessive mutations or de novo variants that might be responsible for the disease. For instance, in a study of autism spectrum disorder (ASD), joint calling across a cohort of affected and unaffected siblings can reveal rare genetic variants associated with the condition, improving diagnostic accuracy and guiding further genetic research.