Result interpretation guide for bacterial whole genome sequencing
In our whole bacterial genome sequencing service we not only deliver you a high quality assembled and polished sequence, but also an informative HTML report.
Oxford nanopore technologies (ONT) sequencing
In ONT nanopore sequencing, the input material directly influences the output. If a flow cell is loaded with small fragment DNA, the resulting reads will be of a corresponding size. Conversely, if high-molecular weight DNA is loaded, longer reads will be obtained. Overall, all molecules included in the sample will be sequenced and the relative read counts of various molecular species will generally align with the actual proportions of those species present in the sample. There is one caveat though: small fragments are sequenced in a higher frequency than longer ones.
Also be aware that agarose gels don’t have a very high sensitivity, only DNA fragments in a high enough concentration can be visualized. There might be more going on at a lower level in the background of the sample (you might see a smear on the gel).
Nanopore sequencing by ONT does not require primers and typically involves sequencing the entire plasmid molecule with read lengths that span its entirety. Therefore, all molecules present in the received sample, including degraded plasmids or background genomic DNA, are sequenced.
Sample requirements:
Our sample requirements for bacterial whole genome sequencing are:
Concentration: 30 ng/µl
Volume: 20 µl
The required DNA concentration according to our specifications is 30 ng/µl. We highly recommend fluorometric concentration measurements (e.g. Qubit) instead of photometric ones (e.g. Nanodrop), because of their significantly higher accuracy for double-stranded DNA. Photometric measurements frequently overestimate the samples’ DNA concentration. We often receive genomic DNA from customers who still measure their concentration with photometric measurements, which is the most common reason for failed attempts of bacterial whole genome sequencing. We will attempt to sequence your sample even when your sample doesn’t fulfill your requirements. We cannot guarantee success, but unfortunately, we must charge for our sequencing attempt .
As our service is optimized for clonal / single bacterial genomes, we recommend controlling the quality of a sample (i.e. a single gel band), preferably as a linearized DNA, on a gel or with a Bioanalyzer/Fragment Analyzer (however watch out for biological concatemers, see below). If your samples failed to be sequenced, please consider performing a new preparation to rule out contamination. Additionally, performing a size selection on a gel could be a good procedure to remove contaminating degraded DNA.
Accuracy of bacterial whole genome sequencing results
According to the specifications provided by Oxford Nanopore for the chemistry and flowcells used in our current bacterial whole genome sequencing, the raw read accuracy exceeds 99%. In general, higher coverage, which refers to having more reads available for consensus building, tends to enhance the accuracy of the results.
Nevertheless, we also deliver a variant calling within the report, which detects positions in the final genome sequence with lower confidence.
Lower confidence bases
Our consensus assembly process utilizes deep sequencing to achieve a high level of accuracy at the individual base level. However, Oxford Nanopore long read data encounters challenges in resolving certain common motifs. To tackle this issue, we polish the sequence to correct many of these problematic bases.
In addition, we employ a strategy where we map your reads against a high-quality consensus assembly to identify lower confidence bases. During this process, we determine the frequency of each nucleotide at a specific position. In regions with high confidence bases, the majority of raw reads will contain the same assembled base. However, in areas that pose challenges, such as motifs like Dcm methylation sites (CC[A/T]GG) or long stretches of homopolymer bases, different nucleotides may be identified at the same position in the raw reads, despite the assembled base potentially being correct.
If your assembly differs from your expectations, it is important to consider these factors.
Errors or low confidence positions in homopolymer region or a Dcm methylation site
Deletions within homopolymer stretches and errors at the middle position of the Dcm methylation sites CCTGG and CCAGG are the most common error modes observed in Oxford Nanopore sequencing.
Sequencing coverage of bacterial genomes
We cannot provide a specific level of coverage guarantee as the number of raw reads generated can significantly vary based on the quality of the sample. Typically, successful samples sent at the recommended concentration yield a substantial number of raw sequencing reads, ranging from high dozens to potentially hundreds or even thousands. The average coverage is indicated in the report, and a coverage of approximately 20x or higher suggests a highly accurate consensus.
For more questions, please also visit our FAQs >>
Data interpretation
Read length histograms
Prior to sequencing your genomes, the library preparation workflow linearizes the DNA to obtain predominantly full-length sequence reads. In the result report we plot a read length histogram, which shows the read length from all DNA molecules present in the sample (and which can be sequenced).
Non-weighted vs. weighted histogram
Distribution of read lengths from sequenced data is shown in the read-length histograms. Read length histograms can be used to assess the quality of sequencing data, as the distribution of read lengths can indicate extraction quality and fragmentation, the presence of contaminants, or biases in the sequencing process.
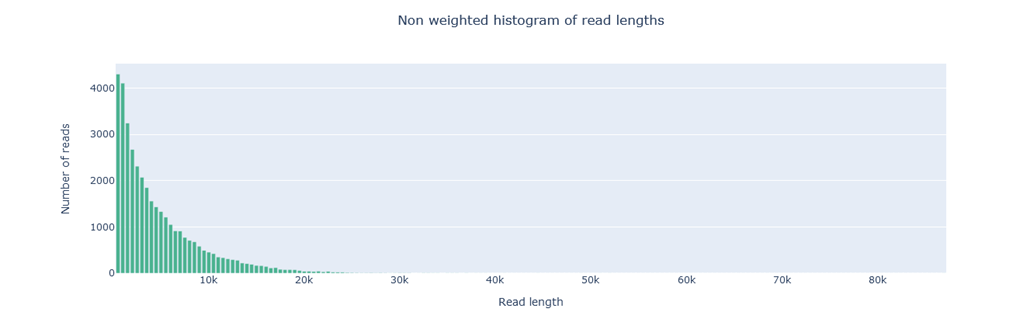
Non-weighted histogram
This histogram displays the number of reads on the y-axis and the read length on the x-axis. Each bar in the histogram represents a range of read lengths, and the height of the bar indicates the total number of reads falling within that range. This histogram is part of the kernel density estimate (kde) plot in our HTML report.
Example of a good result:
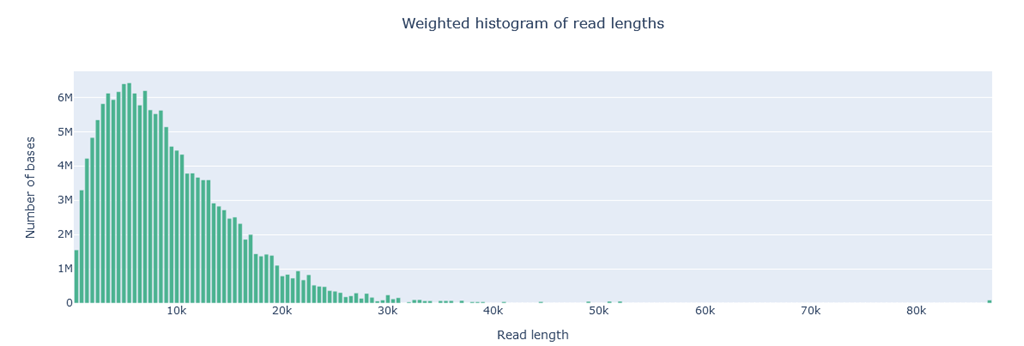
Weighted histogram
The weighted histogram displays the number of sequenced bases (bp) on the y-axis and the read length on the x-axis. Instead of total number reads the height of the bars indicate the total number of bases (bp) falling within that range. This results in a weighted plot by the number of nucleotides per bin, as longer reads carry more weight in the histogram.
Example of a good result: